Services

Genotyping

Metagenome Profiling

Bioinformatics / Analytics
Genotyping
OmeSeq-qRRS is an optimized NGS-based reduced representation sequencing (RRS) platform that circumvents several challenges and limitations associated with NGS-based RRS methods. Initially developed to tackle the difficulties of dosage-sensitive genotyping in complex polyploid genomes, it has since been successfully applied to genotyping a variety of other organisms and metagenome sequencing.
Due to its unique approach and adapter design, OmeSeq offers several key advantages. Improvements include exceptional read quality, a quantitative assay, and uniform sampling across loci and multiplexed samples. These translate to mitigating typical issues such as: (i) high missing data rates, (ii) alleleic dropout, which can lead to under calling heterozygotes and rare alleles, and (iii) barcode swapping/index hopping, which results in the incorrect assignment of sample reads.
Metagenome Profiling
Metagenomics is a powerful tool for understanding organismal interactions within community, ecological, or environmental contexts. While next-generation sequencing has significantly advanced metagenomic community profiling, certain challenges limit the ability to draw functional inferences. These include obtaining strain-level profiling, quantification of community members, high cost, and downstream analyses.
By combining OmeSeq-qRRS with an automated analytical pipeline (Qmatey), quantitative and broad-spectrum taxonomic profiling can be achieved from viruses to eukaryotes. Compared to other metagenomic profiling methods, such as shotgun metagenomic sequencing and amplicon sequencing, OmeSeq-qRRS is very cost-effective (only $15 – $20 per sample from DNA to metagenomic analysis results) and suitable for large scale studies (currently 96- to 9,216-plex level).
Benefits of OmeSeq-qRRS-based Genotyping and Metagenome Profiling
$15/sample (library prep + sequencing) compared to other methods such as deep shotgun (> $1,000), shallow shotgun (~ $500), and amplicon (~ $50) sequencing. | |
~ 1 month | |
$3 per sample | |
Isothermal amplification at a stringent temperature (65℃) and the dsDNA-protection assay prevents off-target hybridization and other factors that drive amplification bias | |
Isothermal amplification and a qPCR-enrichment step for low-input DNA ensures a quantitative assay. | |
Restricting exact matching to taxonomic groups (typically phylum-level) prevents false negatives as endophytic/epiphytic/contaminating microbial sequences tend to be included in genome assemblies. | |
Exact matching at the strain-level is based on 100% sequence identity/uniqueness, while exact matching of consensus sequences (EMC) is applied at 98% (species), 97% (genus), 96% (family), and 95% (order, class, and phylum) identity | |
We employ a novel algorithm that interrogates large databases within hours to days, i.e., tens to hundreds of samples, respectively. It aligns full-length sequences using a fast implementation of MegaBLAST. Amenable to NCBI and custom databases. Given reference fasta sequences, Qmatey can also build (automated) a database for the analyses. | |
Some of the QC steps include: |
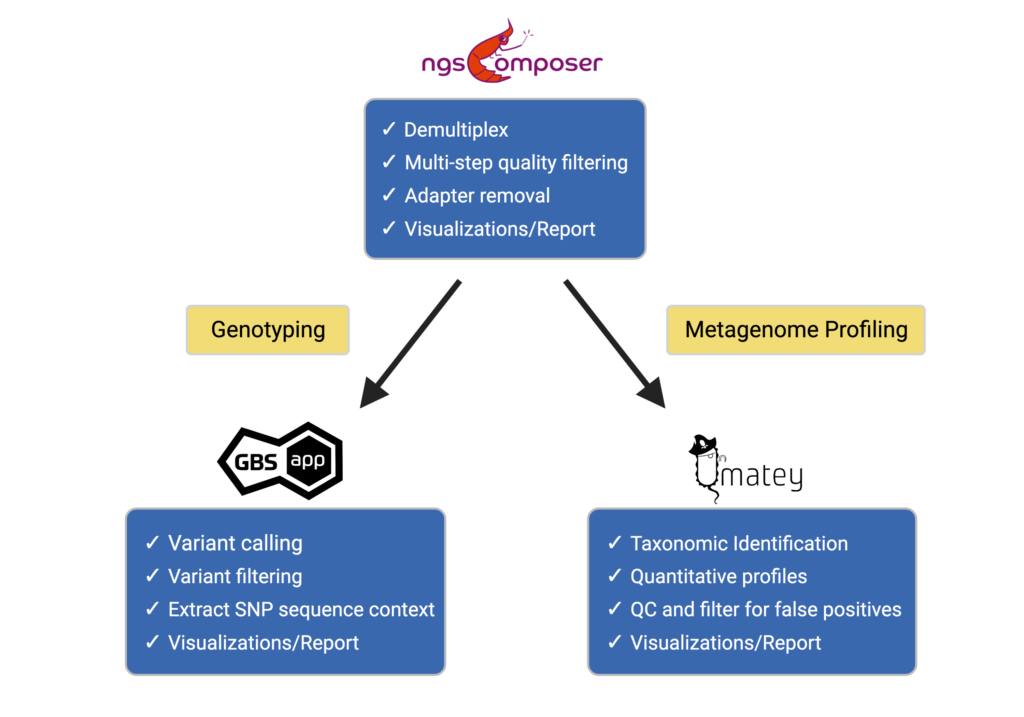
Bioinformatics / Analytics
BASEwise Solutions offers comprehensive data analysis using custom-designed and open-source software that takes into account the novel features of OmeSeq-qRRS. The software integrates both original tools and state-of-the-art open-source third-party tools. The customized software is also compatible with other high-throughput data not generated with the OmeSeq protocol, although results are ultimately dependent on the quality of input data.
Each of the pipelines we employ are fully automated and designed with fast turnaround times (days versus months), which is especially relevant when dealing with large data sets. While all the software is publicly available (see Bioinformatic Tools) and user-friendly for biologists, the computing power required to run these analyses can be limiting without the appropriate infrastructure.
BASEwise Solutions can provide expertise in handling small- and large-scale data sets and offers speedy turnaround times for data analysis. For assistance with project design or implementation, please contact us, or request a free project design consultation.
Bioinformatics Workflow for Genotyping & Metagenome Profiling Data