
Products: Adapters
Adapter Design
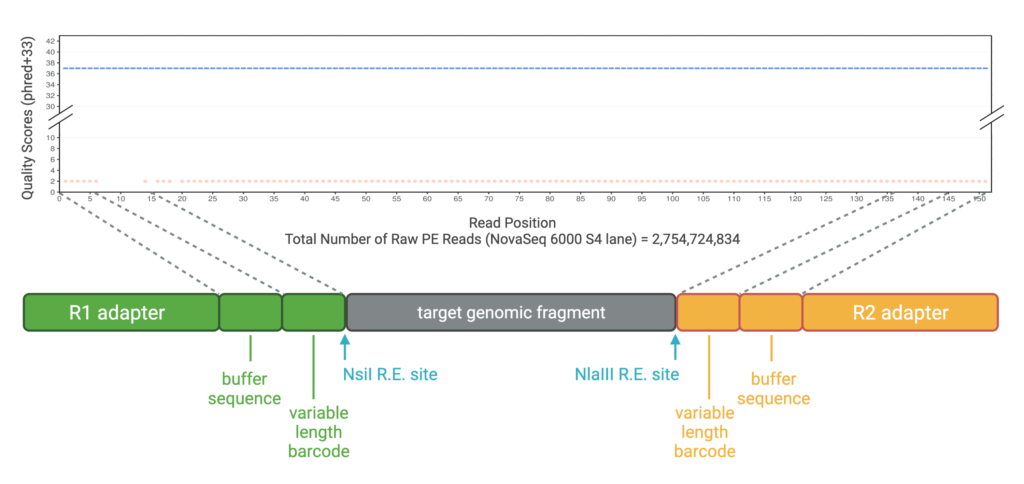
The R1 and R2 adapters used for the OmeSeq-qRRS library construction incorporate features that enhance read quality and allow for scalable multiplexing and genome-wide sampling. In addition to the high-fidelity base calls, complementary bioinformatic tools further ensure that reads are correctly assigned to samples during demultiplexing, i.e., avoiding barcode swapping/index hopping. The R1 and R2 adapters are compatible with all Illumina sequencing platforms.
Adapter Features
Barcode design | Generates high edit/Levenshtein distance between barcode sequences that minimize the chance of barcodes mutating and evading detection. Adapters are rigorously tested to prevent dimerization and motifs that can be problematic for Illumina sequencing. |
Buffer sequence | Ensures barcodes occur in high-fidelity base calling regions. Because the ends of reads are more error prone and often lead to barcode swapping/index hopping, the buffer sequence acts as a spacer to move the barcode further away from the ends. When coupled with novel demultiplexing algorithms in ngsComposer, we can eliminate barcode swapping by an exhaustive search and detection of sequences that could revert to more than one barcode/sample. |
Variable length 96×96 barcodes | Prevents phasing error during sequencing by staggering restriction endonuclease (REase) sequences across samples and ensures nucleotide diversity at each position, a requirement for Illumina sequencing platforms. |
Motif-based error detection and filtering | After barcode removal, we use the presence of error-free REase site motifs as an early indicator of base calling quality across the entire read. Because the REases, NsiI and NlaIII, are used in OmeSeq library preparation, we can use these sequences as a filter for read quality. Empirical studies (Kuster et al. 2021) show that reads that have incorrect base calls in these REase sites tend to propagate along the entire read length, while correct base calls at these REase sites tend to have high quality base calls along the entire read. This is an additional and powerful way to empirically quality filter the data in addition to quality scores. |
Multiplexing with 9,216 adapter pairs (96-R1 x 96-R2 adapters)
The R1 and R2 adapters are provided separately in a 96-well plate format, each containing 96 unique adapters. By combining the adapters in all possible paired combinations, up to 9,216-plex level can be achieved. To ensure optimal nucleotide diversity for high-quality Illumina sequencing, all R1 and all R2 adapters should be used regardless of assay multiplex-level.
The diagrams below indicate the pooling strategies for 96-, 384-, and 1152-plex levels.



Created with BioRender.com